
🔬ESMFold2: The Bitter Lesson is Coming for Proteins - Alex Rives, BioHub
Команда ESM из BioHub (под руководством Alex Rives) выпустила ESMFold2 — открытый научный движок для предсказания, дизайна и исследования белков, построенный на принципах масштабирования языковых моделей. В отличие от AlphaFold2, использующего множественные выравнивания последовательностей (MSA), ESMFold2 опирается на обучение без учителя на 2,8 миллиарда белковых последовательностей, формируя «мировую модель» белков, способную к обобщению. Модель демонстрирует state-of-the-art результаты в предсказании белковых взаимодействий, особенно для антител — критически важной модальности в терапии, — и подтверждает работоспособность масштабирования на этапе инференса по пяти мишеням в онкологии и иммунологии. Вместе с моделью опубликован атлас из 6,8 миллиарда белков и 1,1 миллиарда предсказанных структур, всё — под лицензией MIT. Применение методов механистической интерпретируемости (Sparse Autoencoders) позволяет извлекать семантические признаки, иерархически описывающие структуру белков — от отдельных аминокислот до целых доменов и функциональных мотивов, приближая нас к программируемой биологии.
🔬ESMFold2: The Bitter Lesson is Coming for Proteins - Alex Rives, BioHub
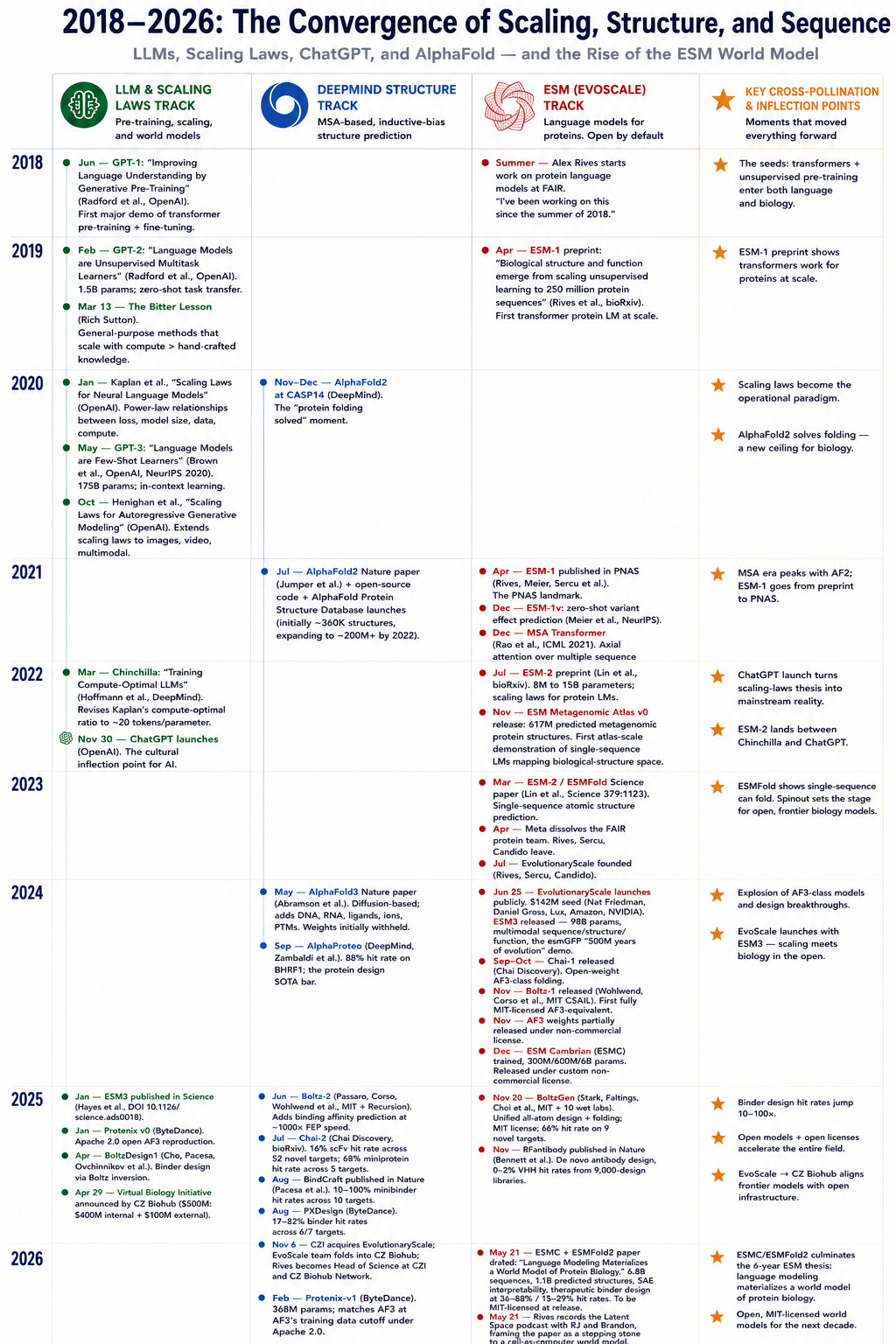
Editor’s note: In our first BioHub pod with Priscilla and Mark they discussed their acquisition of EvoScale, led by Alex Rives, who is now Head of Science at BioHub. With ESM-1 they trained language models on millions of protein sequences drawn from across life, with a simple “next token” objective: predict the amino acids that have been randomly masked out, based on the context of the rest of the sequence. But they soon found that these models also learned biological structure and function, including properties the model had never been explicitly shown AND that this ability scales predictably with compute, leading to ESM2 and ESM3.
Today, Alex announced ESMFold 2, an open scientific engine to power prediction, design, and discovery across protein biology.
Building on Cryo-EM data (discussed in the CZI pod), ESMFold2 reports state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics, and evidence that inference time scaling is also working across five targets in cancer and immunology.
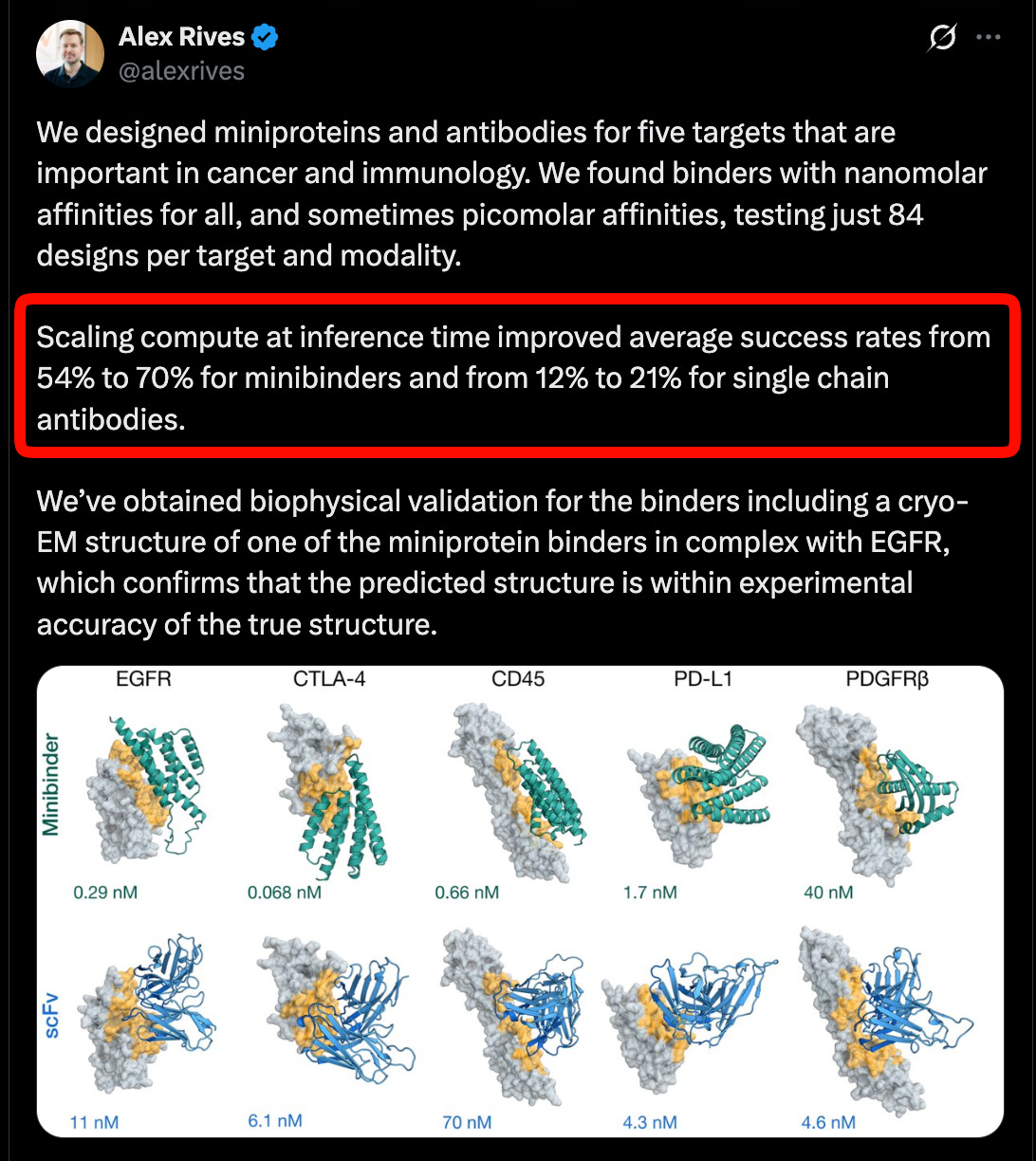
In a nod to that other famous AI x protein folding project, they are also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures, which you can play around with on their website. We are honored to work with them for this huge release!
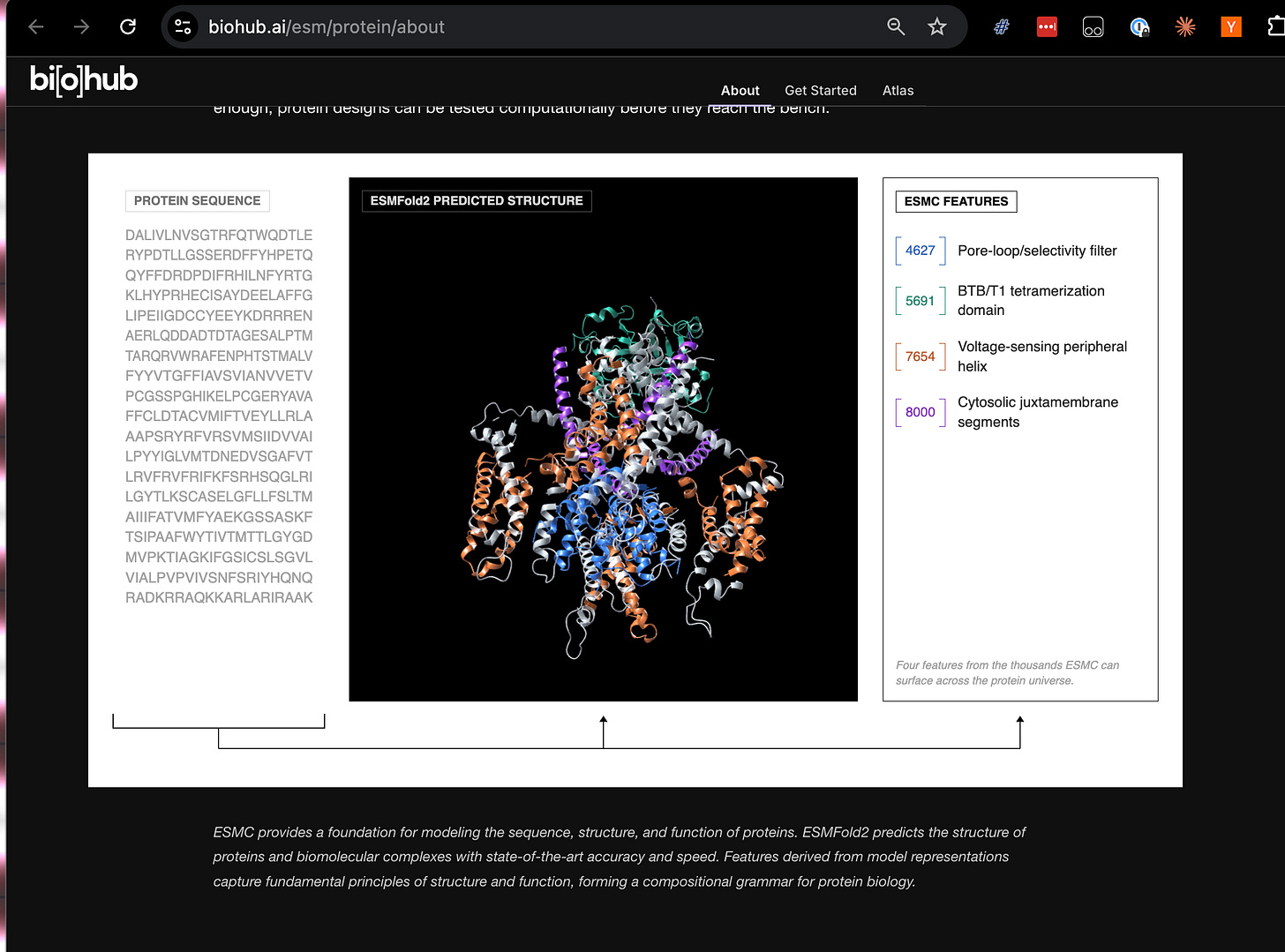
One of the refrains we’ve heard on the Science pod has been that protein folding, materials design, cellular biology, etc. are very different problems from Language Modeling. They definitely are. Yet Alex Rives and the ESM team at BioHub just released a preprint and model, demonstrating that vanilla BERT-like transformer models trained on sufficiently large and diverse data sets can beat specialized models like AlphaFold3 on some of the hardest protein-related problems.
Andrew White had a great segment in our first LS-Science episode that explained how mind blowing AlphaFold2 was when it was released in 2020: it suddenly solved problems on a GPU on your desktop that DESRes had built custom-ASIC supercomputer clusters to solve. John Jumper and Demmis Hassabis received the Nobel Prize in Chemistry for this work.
AlphaFold2 took advantage of an very clever observation: if multiple species co-evolve pairs of mutations, this implies that the mutations correspond to parts of the protein that are close in 3d space. This is usually shorthanded as MSAs (multi-sequence alignments), and is the key insight which makes AlphaFold2 so effective.
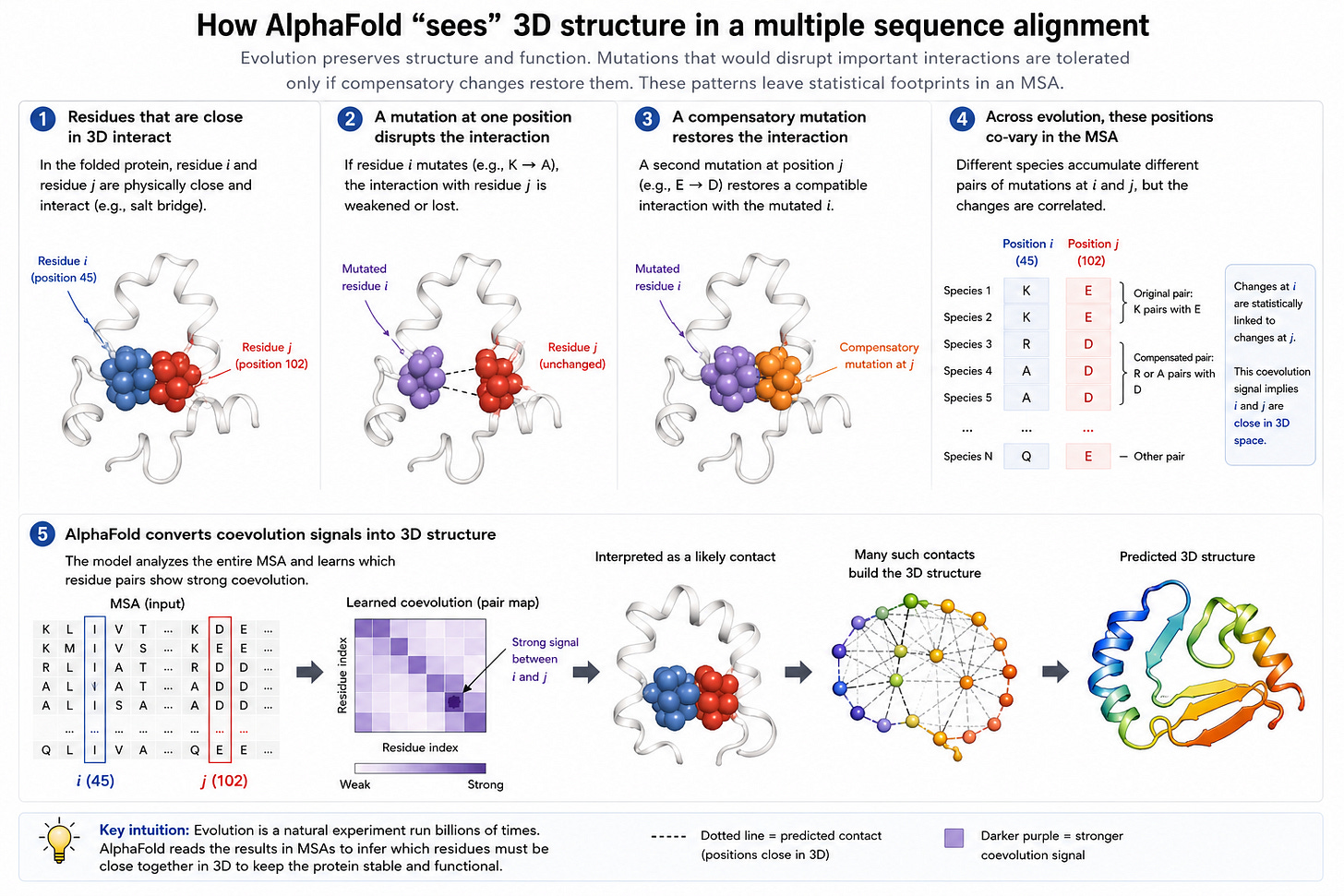
Like other inductive biases, however, it hurts generalization.
Scale-pilled before it was cool
If you take a look at the timeline for scaling laws for LLMs and release of structure prediction models1, the ESM team notably doubled down on their MSAs-be-damned approach after AlphaFold2 released. This obviously requires a great deal of belief in the scale hypothesis.
Why the conviction?
ESM developed at a time when many of the scaling laws and the “Bitter Lesson” were proving increasingly correct. AlphaFold2’s wild success must have been both exciting and bitterly disappointing. But using MSAs mean that the model is is dependent on training data that contains MSAs in order to be accurate in a given domain. For things like antibodies that don’t have MSAs to train on2, AlphaFold tends to do poorly.
ESM takes a different approach: learn the relationship between different proteins by unsupervised training on as much diversity as you can find (sound familiar?) and then correlate that back to structures know from the Protein Data Bank (PDB) and other sources3.
In other words, a World Model.
World Model for proteins
“World Model” is a hype term that I define like this:
Use unsupervised training to learn abstract patterns from the data:
The abstraction should be semantic - novel constructions represent things that obey the rules of the real world
The abstraction should be compositional - recombining different patterns leads to novel and often valid constructions
The abstraction should support generalization - it predicts things in the real world it wasn’t trained on
Once you have a world model, you can attach “heads” to it for downstream tasks: predict properties of a protein, decompose its functional features, or search the representation for proteins that meet design criteria. The two big models BioHub just released under MIT license map directly onto this:
World model → ESMC (a model trained on 2.8 billion sequences)
Structure-prediction head → ESMFold2
One of the interesting ways the world model can “predict things” is to generate proteins sequences and then measure the predicted properties, such as binding affinity, in the lab. Alex talks in the episode about validating some of the harder molecules they predicted in the wet-lab. Very cool!
Another way is to use mech-interp techniques such as Sparse Auto Encoders (SAEs) to extract semantic features from your model, and then find novel features that predict unknown biology. I won’t spoil this part for you: it was one of the highlights of the episode for me!
A cell is a computer
We have all heard that genes are like computer programs, but usually the analogy fizzles after that. Of course genes are transcribed into RNA and RNA is translated into proteins, so genes are programs for building proteins, but that carries the analogy only to “binary digits are programs.”
Here’s a better analogy: you can think of the cell nucleus as a storage device / storage controller, the ribosome as a JIT-compiler and runtime, and the semantic features that we learn from our world model via SAEs as functions, proteins as processes that interact together in workflows (signalling pathways) to produce behaviors and outputs (phenotypes).
Like functions, the SAE features have a hierarchical composition from local, secondary and tertiary structures (mimicing protein structure)4, but also motifs that are conceptual, such as membrane integrations, disordered regions and disulfide bonds5. As we learn to compose these features we into novel protein designs, we move further towards programmable biology.
Alex goes into much more detail about this in the episode, as well as:
Principles for new data collection
BioHub’s vision
Modeling the cell
Enjoy!
Full Video podcast
please like and subscribe!
LinkedIn:
Antibodies mutate very rapidly so that they can adapt to pathogens with novel proteins on them. These dynamics mean that MSAs don’t appear in them.
This includes a dataset created using AlphaFold2 itself for ESMC, making it a distillation of AlphaFold, and indirectly dependent on MSAs itself.
Very local (1–3 residues): individual amino acid biochemistry, hydrophobic vs. polar character, charge
Short-range (~5–10 residues): secondary structure — α-helix features, β-strand features, β-turn features
Medium-range (~10–30 residues): supersecondary motifs — β-hairpins, helix-turn-helix, β-α-β units
Long-range (whole-protein): full domain identifiers — immunoglobulin fold, Rossmann fold, TIM barrel, four-helix bundle
DNA-binding features — activated across helix-turn-helix proteins, zinc fingers, leucine zippers, and other DNA-binding folds that share function but not sequence
Membrane integration features — activated on transmembrane segments regardless of whether they sit in a GPCR, a transporter, or a channel
Disordered region features the SAE devotes ~686 features (5–10% of the feature budget) to intrinsically disordered regions, which is striking because IDRs have no structure to predict. The model represents disorderedness itself as a concept, with sub-features for different IDR flavors (polyampholyte, polar tract, prion-like domain)
Disulfide bond features — activated on cysteines that participate in disulfides, distinguishing them from free cysteines
Discussion about this episode







